Klinische Studien
Das Arzneimittelgesetz (AMG) unterscheidet zwei Kategorien: Nicht-Interventionelle Studien (NIS, vormals Anwendungsbeobachtungen) und Klinische Prüfungen.
Um eine NIS durchführen zu dürfen, muss das Arzneimittel zugelassen sein und streng nach den Vorgaben dieser Zulassung angewendet werden. Weiters darf es keine Untersuchungen außerhalb der Routine oder sonstige Belastungen für die Patienten geben. Im Gegensatz dazu können in der klinischen Prüfung auch nicht zugelassene Arzneimittel oder Arzneimittel außerhalb der Zulassung erforscht werden, und es sind zusätzliche Untersuchungen erlaubt.
Beide Formen dienen der Beantwortung von wissenschaftlichen Fragestellungen.
Klinische Prüfungen von Arzneimitteln unterliegen neben dem Arzneimittelgesetz (AMG) der europäischen Verordnung (EU) 536/2014 ("Clinical Trials Regulation"). Die gesetzlichen Vorgaben sind daher im gesamten EU-Raum dieselben. Weitere Informationen zur Genehmigung von Klinischen Prüfungen finden Sie hier.
Einteilung in Phasen
Interventionelle Studien werden in Phasen eingeteilt, Phase I stellt die früheste Phase dar und beinhaltet auch Studien mit Erstanwendung am Menschen. Während in der Phase I primär auf mögliche unerwünschte Wirkungen des Arzneimittels geachtet wird, untersucht man in Phase II bereits mögliche therapeutische Effekte. Phase III Studien werden auch als Pivotalstudien bezeichnet, da das Ergebnis dieser Studien ausschlaggebend für spätere Zulassungen ist. Phase IV Studien beschreiben die Untersuchung von bereits zugelassenen Arzneimitteln, wenn die zugelassene therapeutischen Anwendung um zusätzliche Untersuchungen erweitert wird.
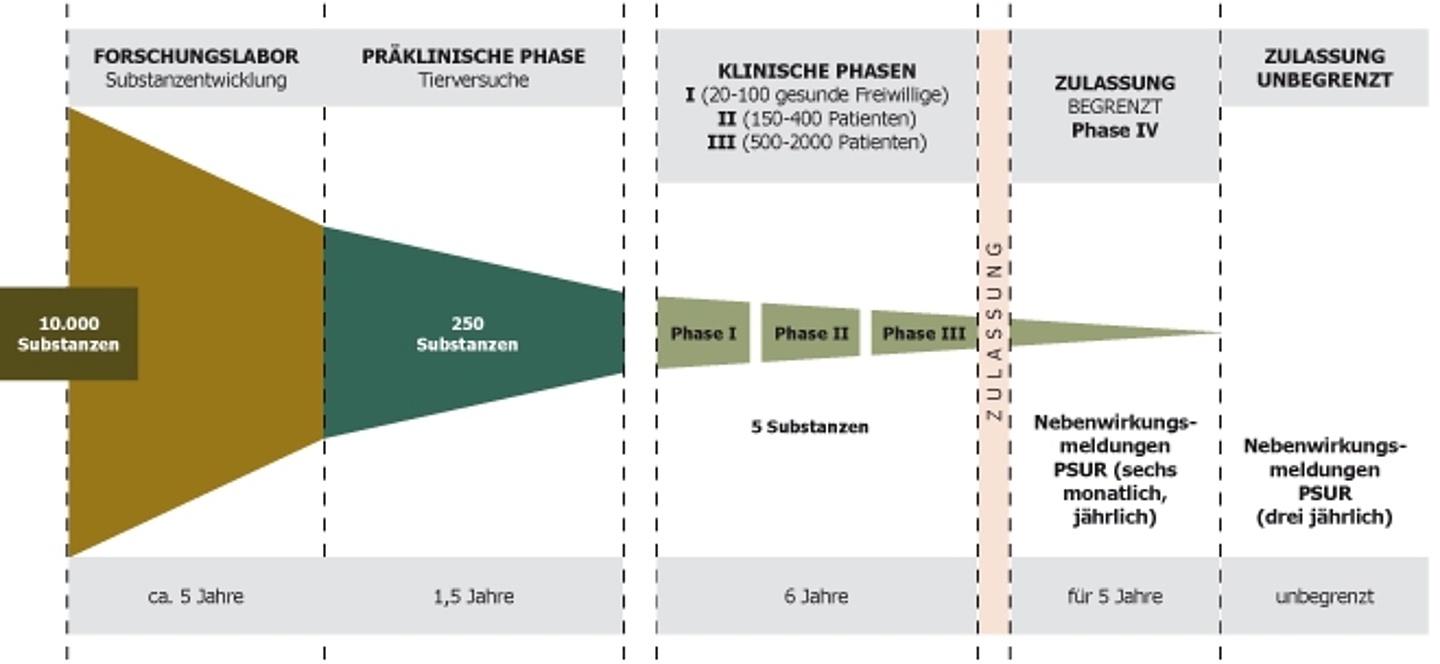
Regulatorische Voraussetzungen für die Durchführung von Arzneimittelstudien
Interventionelle klinische Arzneimittel-Prüfungen müssen sowohl von der Behörde (BASG), als auch von den Ethikkommissionen genehmigt werden.
Die Einreichunterlagen bestehen aus folgenden drei Teilen:
- Angaben zur Herstellung, Stabilität und Reinheit des Prüfpräparates
- Angaben zur Pharmakologie und Toxikologie (primär Daten von Tierstudien)
- Ergebnisse bisheriger klinischer Prüfungen, Studienprotokoll und Patientenaufklärung.
Diese Voraussetzungen für die Genehmigung von Interventionellen Arzneimittelstudien sind in Europa einheitlich geregelt. Klinische Arzneimittelstudien von neuen Arzneimitteln ohne vorherige Tierstudien gibt es (normalerweise) nicht. Meist liegen bereits jahrelange präklinische Erfahrungen mit dem Arzneimittel vor, bevor überhaupt mit Phase I Studien gestartet wird.
Die Behörden und die Ethikkommissionen üben eine wichtige Kontrollfunktion aus, um für vergleichbare Standards bei Arzneimittelstudien und für eine möglichst hohe Patientensicherheit zu sorgen.
Rechte des Studienteilnehmers
Patienten, die an einer Studie teilnehmen, müssen der Teilnahme explizit zustimmen. Diese Zustimmung erfolgt nach einer sorgfältigen mündlichen Aufklärung durch den Prüfer und einer gründlichen Durchsicht der Patienteninformation sowie der Unterzeichung der Einwilligungserklärung.
Detaillierte Kontaktinformationen sind ein wesentlicher Bestanteil der Patienteninformation. Im Falle von Unklarheiten muss dem Patienten die Möglichkeit gegeben werden, sich an eine Kontaktperson (z. B. an den Prüfarzt oder die Patientenanwaltschaft) zu wenden.
Der Patient kann jederzeit ohne Angabe von Gründen und bei weiterhin bester medizinischer Betreuung aus einer klinischen Prüfung aussteigen.